Theoretical Studies on Organometallic Catalysis
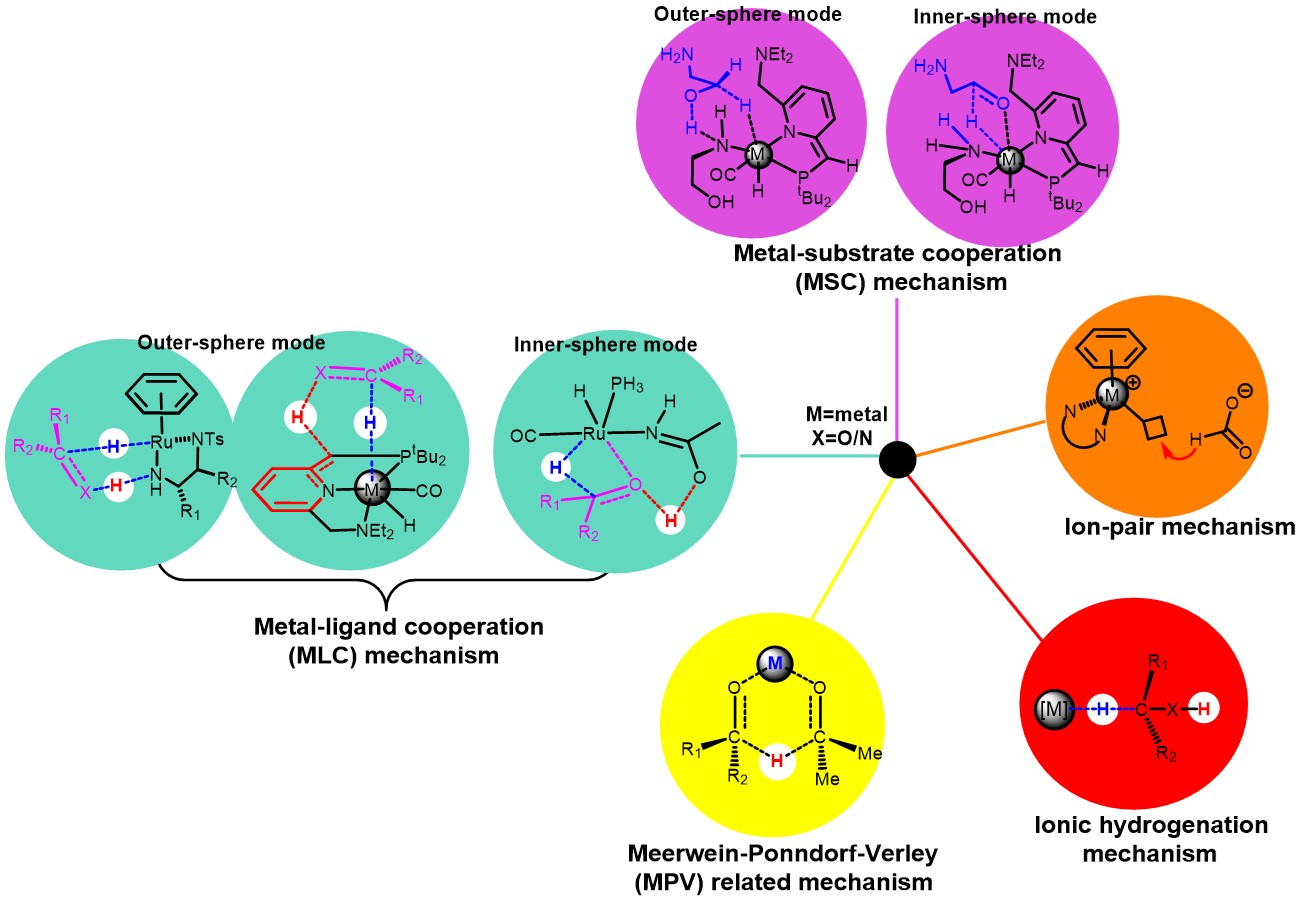
By means of theoretical and computational chemistry, we investigated reaction mechanisms of small molecule activation, C-H bond activation and asymmetric reaction catalyzed by organometallic complexes systematically, including the activation of dihydrogen, nitrogen, carbon dioxide and olefins et al. Some fundamental reactions such as substitution, migration and insertion, hydrogen transfer, oxidative addition/reductive elimination, metathesis and nucleophilic addition were discussed. The mechanisms of organometallic catalytic reactions were summarized to unveil the nature of reaction activity and selectivities. The factors like solvent effect on organometallic catalysis were considered. Meanwhile, the three-dimensional quantitative structure performance relationship (3D-QSPR) model was established, which could promote the design and discovery of new skeleton transition metal catalyst with high activity and enantioselectivity based on the reaction mechanism (This is supported by National Natural Science Foundation of China (Grant nos: 21672018, 2161101308, 21373023, 21072018) and Beijing Municipal Natural Science Foundation (Grant nos: 2242014, 2162029)).
Theoretical Studies on Catalysis on Surfaces
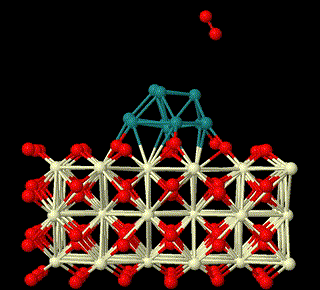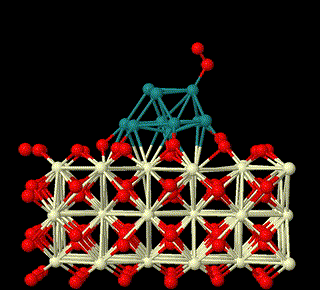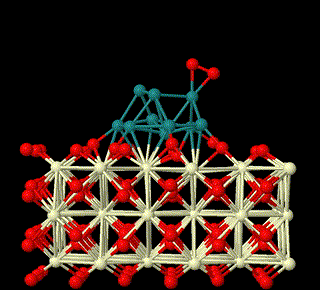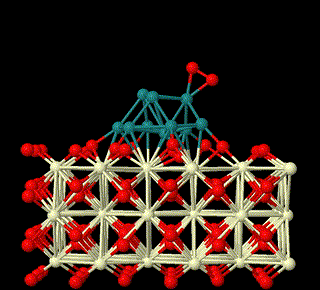
The first-principles theory and DFT method are used to study the heterogeneous catalysis on surfaces. Here we focus on reaction mechanisms of catalysis on surfaces involving metal oxides related to small molecules activation in sustainable energy field, to unveil the nature of heterogeneous catalysis and build relationship between structures of surfaces and their catalytic activities (This is supported by National Natural Science Foundation of China (Grant nos: 22473008, 22411530047, 22073005).
Computational Biophysical Chemistry
The three-dimensional structure and dynamics are closely related with the function of biomolecules. By means of QM/MM, molecular dynamic simulation, docking and 3D-QSAR methods, the relationship between the dynamics and the function of the molecule and biocatalysis mechanism at atomic level were investigated, which be very useful to design new drugs on biomolecular targets (This is supported by NSFC grant No: 20703003).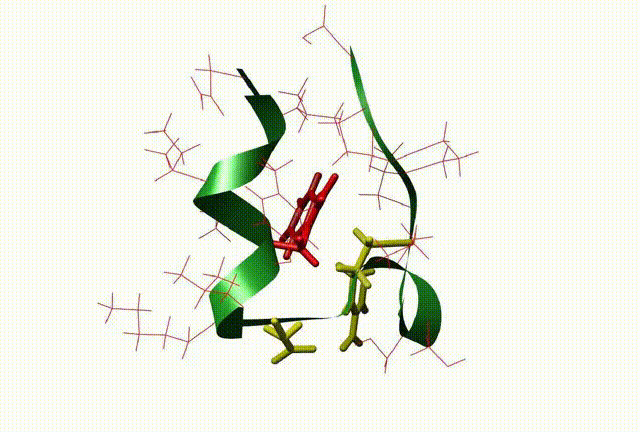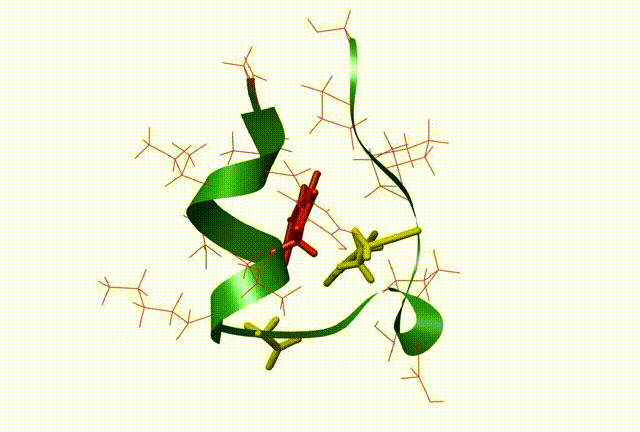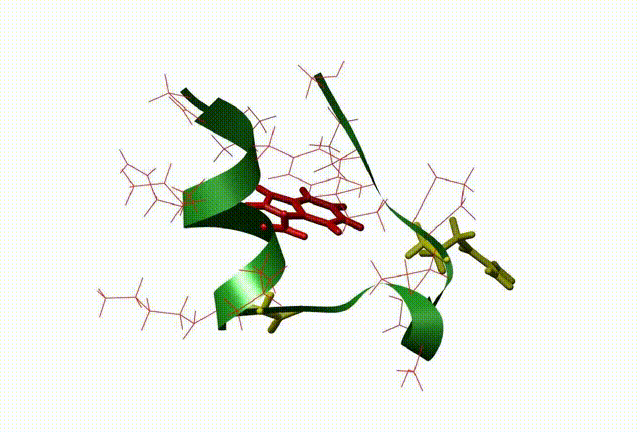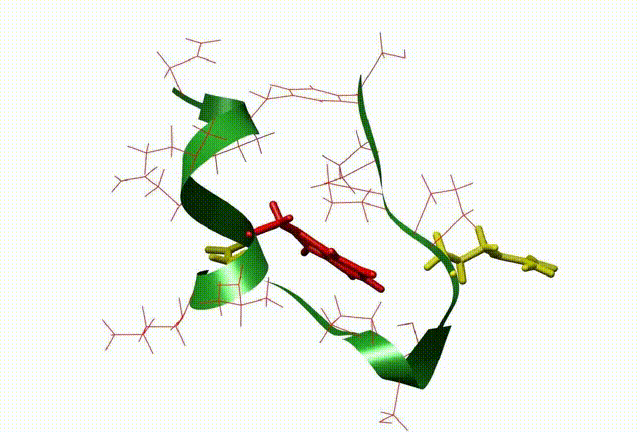
J Software Development
J project aims on the development of a GUI software to construct molecules, display the model, investigate reaction mechanism, perform data mining, build QSAR relationship. This will be used to develop new catalysts based on reaction mechanisms. In addition, some education functions will also be developed such as point group conception.
 |
 |
_____________________________________________________________
Research support
The researches in our group are supported in part by grants from the following agencies:
National Natural Science Foundation of China (NSFC Grant No. 22473008 (2025-2028, PI M Lei), 22073005 (2021-2024, PI M Lei), 21672018 (2017-2020, PI M Lei), 21373023 (2014-2017, PI: M Lei), 21072018 (2011-2013, PI: M Lei), and 20703003 (2008-2010, PI: M Lei))
The Joint Research Fund of National Natural Science Foundation of China (NSFC) and Academy of Finland (AF) (NSFC-AF Grant No. 22411530047 (2024-2026, PI: M Lei))
The Joint Research Fund of National Natural Science Foundation of China (NSFC) and Royal Society of UK (RS) (NSFC-RS Grant No. 2161101308 (2017-2019, PI: M Lei))
The Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase) (2015-2017, PI: M Lei)
Beijing Municipal Natural Science Foundation (BJNSF Grant No. 2242014 (2024-2026, PI: M Lei), 2162029 (2016-2018, PI: M Lei))
Open Research Fund of State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen Univ. (Grant No. 201811, 2019-2020, PI: M Lei)
Open Research Fund of State Key Laboratory of Natural and Biomimetic Drugs in Peking Univ. (ORF of SKLNBD Grant No. K20100103, 2009-2011, PI: M Lei)
Fundamental Research Funds for the Central Universities, MOE (FRFCU Grant No. PYCC1708 (2017, PI: M Lei), XK1527 (2015-2016, PI: M Lei), ZZ1020 (2008-2010, PI: M Lei))
NHwa Pharmaceutical Corp. Foundation (NHwa, 2006-2008, PI: M Lei)
Scientific Research Foundation for the Returned Overseas Chinese Scholars, MOE (SRF for ROCS, 2006-2008, PI: M Lei)
Beijing Nova Fund (2005B17, 2005-2008, PI: M Lei)
 |
 |
 |
 |
 |